Un tournant méthodologique pour l’étude des maladies anciennes
Les maladies infectieuses ont eu des effets dévastateurs sur les populations humaines tout au long de l’histoire, mais d’importantes questions subsistent quant à leurs origines et à leurs dynamiques passées.
Jusqu’à récemment, l’histoire des maladies infectieuses reposait principalement sur des sources écrites, archéologiques ou paléopathologiques. Mais ces approches ont leurs limites : seules quelques maladies comme la lèpre ou la tuberculose laissent des marques visibles sur les os, et les textes anciens sont souvent ambigus ou incomplets. Grâce aux avancées dans les techniques d’ADN ancien (aDNA), il est désormais possible de récupérer des preuves génomiques directes des infections microbiennes passées et de reconstruire des génomes complets de pathogènes anciens : c’est ce qu’on appelle l’archéogénomique.
Dans une étude colossale, publiée en mai 2025 dans Nature, une équipe de chercheurs internationale a recouru à la métagomique shotgun. C’est une méthode de biologie moléculaire qui permet de détecter de manière exhaustive et non ciblée tous les agents microbiens (bactériens, viraux, fongiques, parasitaires) dans un échantillon biologique, y compris ceux encore inconnus. Elle consiste à fragmenter de façon aléatoire (fragmentation shotgun) l’ensemble des acides nucléiques présents dans l’échantillon biologique. Tous les petits fragments, issus aussi bien de l’hôte que des microorganismes, sont ensuite séquencés et analysés grâce à des outils bioinformatiques pour exclure les séquences humaines. Les données restantes sont comparées aux génomes présents dans les bases de données afin d’identifier les agents pathogènes.
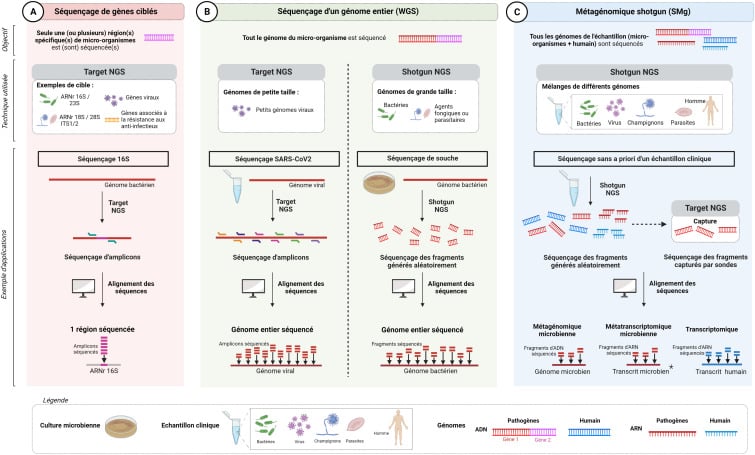
Source : Marchand Sarah et al. (2024) https://doi.org/10.1016/j.revmed.2023.05.002
Les chercheurs ont appliqué cette technique à des échantillons humains anciens (dents, os) provenant de 1 313 individus, répartis sur 37 000 ans et sur une vaste zone allant de l’ouest de l’Europe jusqu’à l’Asie du Sud-Est. Leur objectif : dresser un inventaire moléculaire des pathogènes ayant affecté les populations anciennes, en se basant sur des traces d’ADN microbien retrouvées dans les restes humains.
Criblage métagénomique et analyse bioinformatique à grande échelle
La première étape a consisté à cribler les données de séquençage shotgun. L’équipe a ainsi analysé près de 405 milliards de lectures (reads) de séquences, en appliquant un pipeline bioinformatique haute performance.
Pour distinguer l’ADN pathogène ancien des nombreuses séquences d’origine environnementale (sol, bactéries du nécrobiome), les chercheurs ont mis en œuvre des techniques rigoureuses d’authentification de l’ADN ancien :
- Estimation des dommages de l’ADN (aDNA damage) : l’ADN ancien est caractérisé par des motifs spécifiques de dégradation (comme la désamination en position terminale). Les séquences présentant un score de dégradation (Z ≥ 1.5) ont été retenues comme authentiques.
- Comparaison avec des bases de données modernes : les fragments ont été alignés avec les génomes de 492 espèces pathogènes connues. La similarité moyenne (ANI – Average Nucleotide Identity) a permis de mesurer la proximité des fragments anciens avec les souches modernes.
- Contrôle des artefacts de mélange : les chercheurs ont quantifié le taux d’allèles multiples (multi-allele rate) pour détecter la présence de plusieurs souches ou espèces dans un même échantillon, indicateur possible de contamination ou de co-infection.
Résultat : 5 486 occurrences de pathogènes ont été détectées, dont 3 384 appartenant à des espèces pathogènes humaines bien caractérisées. La majorité ont été identifiées dans des dents, un tissu propice à la conservation de l’ADN circulant dans le sang, et provenaient probablement du microbiome oral endogène (Actinomyces a été retrouvé dans près de 30 % des échantillons et Streptococcus dans près de 20 %). Les autres espèces correspondaient pour une grande partie à des espèces que l’on trouve couramment dans le sol, tels que Clostridium (près de 20 % des échantillons) et Pseudomonas (près de 10 %).
De la peste au paludisme : la répartition des pathogènes anciens
Parmi les multiples germes pathogènes isolés, les chercheurs ont identifié 42 cas putatifs de peste à Yersinia pestis (dont 35 nouvellement rapportés), dont trois datent de 5 700 à 5 300 ans avant le présent (AP), bien avant les grandes pandémies historiques, et s’étendent sur une large zone géographique allant de l’ouest de la Russie à l’Asie centrale et au lac Baïkal en Sibérie. Une découverte qui remet en question les interprétations précédentes selon lesquelles les souches de peste anciennes ne représentent que des spillovers zoonotiques isolés. L’équipe a également documenté des occurrences de peste remontant au néolithique tardif et à l’Âge du Bronze (2 500 AP) dans les steppes eurasiennes, et a mis en évidence plusieurs cas où plusieurs personnes, inhumées simultanément, avaient contracté l’infection. Ces résultats indiquent que la transmissibilité et le potentiel d’épidémies locales étaient probablement plus élevés que ceux supposés précédemment. Sur les 42 cas étudiés, 11 ont été identifiés à la fin des périodes médiévale et moderne (800–200 ans AP) dans deux cimetières du Danemark (Aalborg, Randers), ce qui reflète la prévalencePrévalence Nombre de personnes atteintes par une infection ou autre maladie donnée dans une population déterminée. de la peste durant ces périodes en Europe. Certaines lignées présentaient déjà des gènes de virulence (comme le plasmide pCD1), tandis que d’autres étaient dépourvues du gène ymt, crucial pour la transmission par les puces, suggérant des modes de propagation différents au néolithique.
Borrelia recurrentis, responsable de la fièvre récurrente à poux (dont le taux de mortalité atteint jusqu’à 40 %), serait en réalité apparue dès le néolithique en Scandinavie. Détectée chez 34 individus d’Europe, d’Asie centrale et de Sibérie, cette maladie transmise par les poux de corps a connu des pics de fréquence pendant l’Âge du Fer et l’époque viking, associés à des conditions de vie surpeuplées, à une mauvaise hygiène et à des saisons humides et froides.
Mycobacterium leprae, agent de la lèpre, ne daterait que de l’Âge du Fer tardif, principalement en Scandinavie. L’étude corrobore l’hypothèse selon laquelle le commerce de fourrures, notamment de l’écureuil roux, a pu favoriser sa diffusion zoonotique.
Plasmodium vivax, parasite du paludisme, a été identifié dans des échantillons datant de l’Âge de bronze à la période médiévale, notamment en Europe de l’Est. Il s’agit de la première détection paléogénomique du paludisme dans ces régions.
Les chercheurs ont également détecté 28 cas d’infection par le virus de l’hépatite B (VHB) ; parmi ceux-ci, certains dataient du Mésolithique sibérien (~10 000 AP), étendant considérablement la chronologie connue de ce virus.
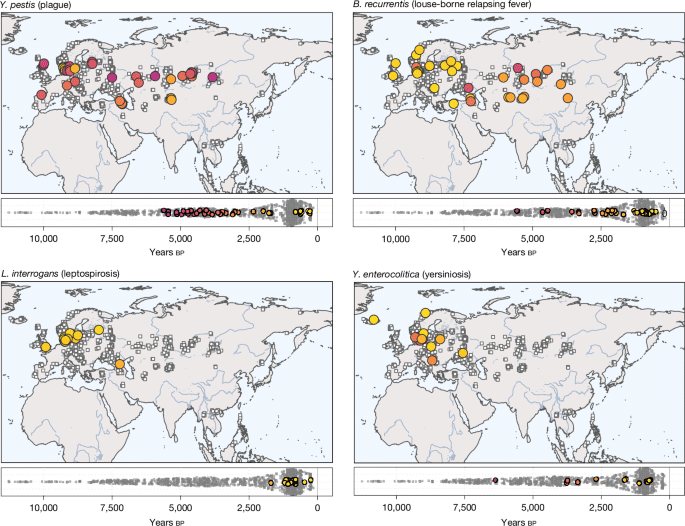
Distribution spatio-temporelle des agents pathogènes anciens sélectionnés.
Chaque panneau montre la distribution géographique (en haut) et la chronologie (en bas) pour les cas identifiés du pathogène respectif (indiqué par des cercles colorés). Les emplacements géographiques et les distributions d’âge de tous les n = 1 313 échantillons d’étude sont présentés dans chaque panneau à l’aide de carrés blancs.
Source : Sikora Martin et al. (2025) https://doi.org/10.1038/s41586-025-09192-8
Nouvelles espèces détectées
Parmi les espèces nouvellement rapportées grâce aux données paléogénomiques, les auteurs mentionnent 12 cas putatifs de yersiniose à Yersinia enterocolitica, couramment contractée par la consommation de viande contaminée crue ou insuffisamment cuite (sangliers, cerfs, chevaux, bovins et moutons). Comme Y. enterocolitica entre rarement dans la circulation sanguine, les données obtenues par l’équipe sous-estiment probablement la charge de la maladie. Cette espèce semble responsable des infections zoonotiques supposées les plus anciennes identifiées par l’équipe chez des individus chasseurs-cueilleurs du mésolithique (Danemark, 6 446–6 302 AP).
D’autres entérobactéries de transmission oro-fécale ont été détectées, notamment des genres Shigella, Salmonella et Escherichia. L’équipe rapporte aussi les premières preuves de leptospirose ancienne datant du néolithique (Suède, Leptospira borgpetersenii). Alors que les cas antérieurs impliquaient principalement L. borgpetersenii (n = 5), la plupart des détections rapportées par l’équipe sont des Leptospira interrogans (n = 20), presque exclusivement en Scandinavie à partir de la période Viking. Aujourd’hui, on trouve principalement L. borgpetersenii chez les bovins, tandis que L. interrogans est détectée plus largement chez les animaux domestiques et sauvages. Bien que les manifestations cliniques soient similaires, les voies de transmission diffèrent – plutôt d’hôte à hôte pour L. borgpetersenii, et davantage via l’urine contaminée pour L. interrogans.
L’équipe rapporte également 2 cas putatifs de Corynebacterium diphtheriae, l’agent causal de la diphtérie, dont le plus ancien remonte au mésolithique.
Une dynamique temporelle liée aux changements culturels
En analysant l’incidence des pathogènes dans le temps, les auteurs ont observé une absence quasi totale d’infections zoonotiques datant d’avant 6 500 AP. L’apparition de l’agriculture et de l’élevage a entraîné une augmentation marquée des zoonoses, avec un pic autour de 5 000 AP, période de grandes migrations pastorales en provenance des steppes.
L’augmentation des contacts interhumains (liés à la sédentarité et à l’urbanisation) et inter-espèces (liés à la domestication), a probablement favorisé le passage d’agents pathogènes des animaux vers l’homme — un mécanisme à l’origine de plus de 60 % des maladies émergentes modernes.
Coinfections, immunité et sélection naturelle
L’analyse a aussi mis en évidence plusieurs cas de coinfections, notamment chez un individu viking porteur à la fois de la variole et de la lèpre. Les chercheurs suggèrent que ces associations reflètent un état d’immunosuppression ou de transmission croisée dans des environnements à forte promiscuité.
Enfin, l’étude éclaire les liens entre pression infectieuse et évolution génétique. Les pasteurs des steppes, exposés de façon prolongée aux animaux, auraient probablement acquis une immunité partielle contre certaines zoonoses, facilitant ainsi la diffusion de ces maladies à l’ouest et à l’est lors de leurs déplacements. Ces dynamiques pourraient avoir contribué au changement génétique observé en Europe, notamment par le biais d’épidémies zoonotiques ayant provoqué le déclin des populations locales, suivies du repeuplement des zones affectées par de nouveaux arrivants qui se sont métissés avec les résidents restants. Les auteurs suggèrent que la pression pathogène aurait entraîné une sélection positive de gènes immunitaires chez les populations des steppes (variants génétiques aujourd’hui associés à des maladies auto-immunes comme la sclérose en plaques), après le début de l’Âge du Bronze en Europe.
Vers une paléo-épidémiologie moléculaire intégrée
Au-delà de l’inventaire des pathogènes, cette étude marque un tournant méthodologique. Elle démontre la faisabilité d’une paléo-épidémiologie de grande échelle, intégrant données génomiques, archéologiques, climatiques et anthropologiques, et comment cette nouvelle discipline peut aider à représenter la distribution spatiale et temporelle de divers agents pathogènes humains au cours des millénaires.
Notre carte actuelle montre clairement que les changements de mode de vie au cours de l’Holocène ont entraîné une transition épidémiologique, qui s’est traduite par une augmentation du fardeau des maladies infectieuses zoonotiques. Cette transition a profondément affecté la santé et l’histoire de l’homme au cours des millénaires. Preuve que les changements anthropologiques peuvent modifier en profondeur l’écologie des maladies — un enseignement précieux dans le contexte actuel d’émergences infectieuses liées aux bouleversements environnementaux et aux interactions accrues entre humains, animaux et écosystèmes, qui souligne, s’il en était encore besoin, de l’intérêt de la démarche One Health.
Référence
Sikora M. et al., The spatiotemporal distribution of human pathogens in ancient Eurasia, Nature, 2025. DOI : 10.1038/s41586-025-09192-8